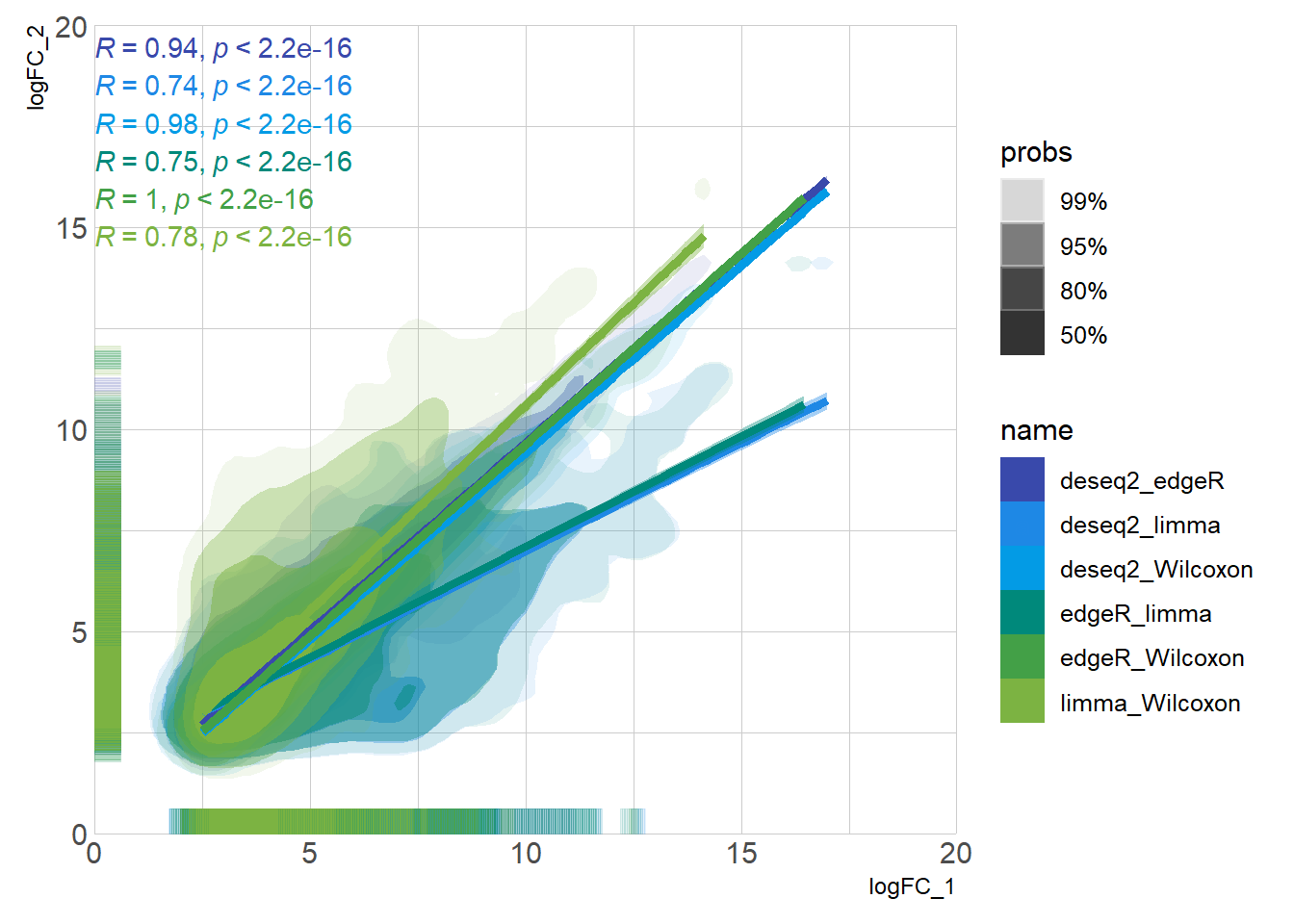
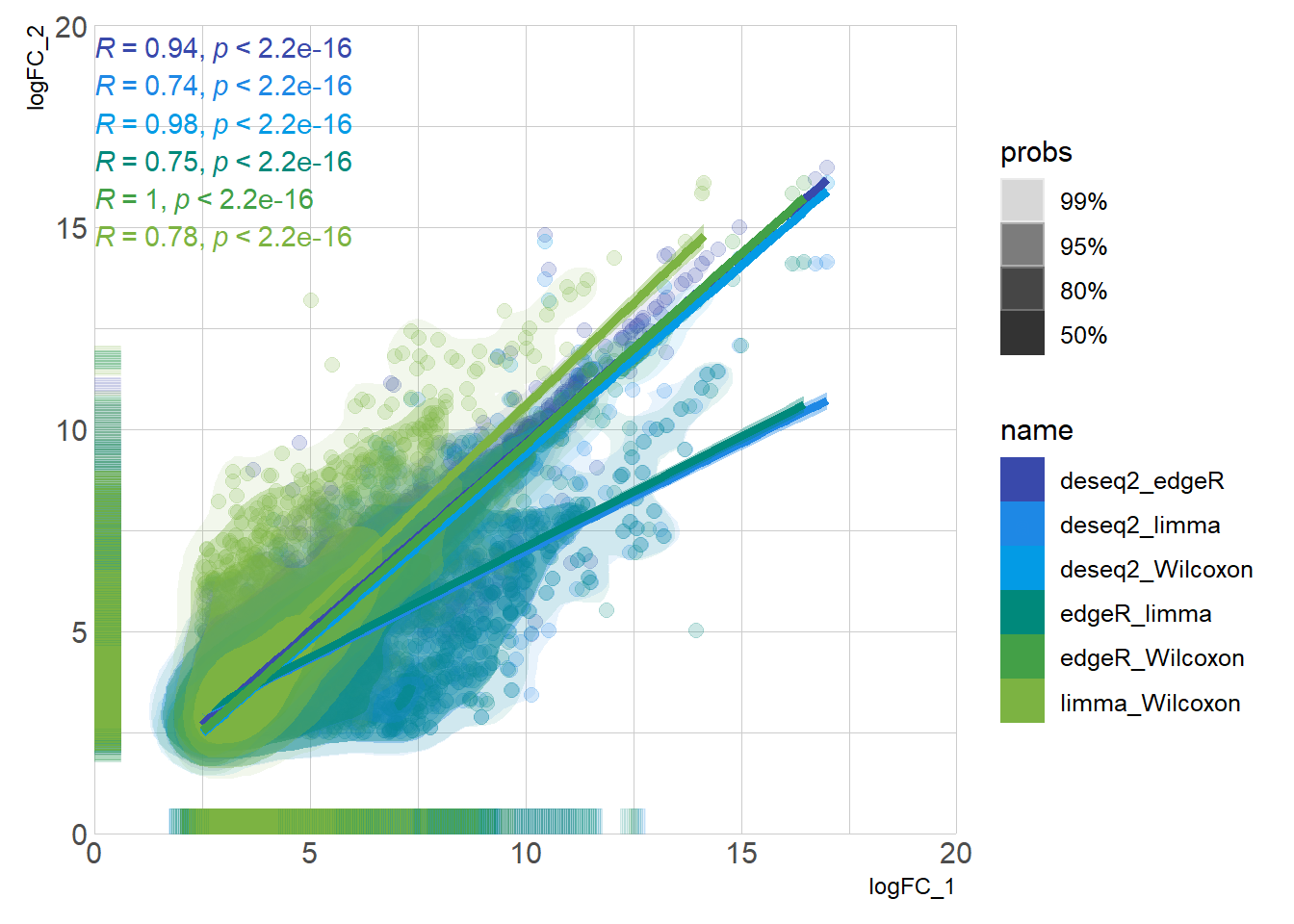
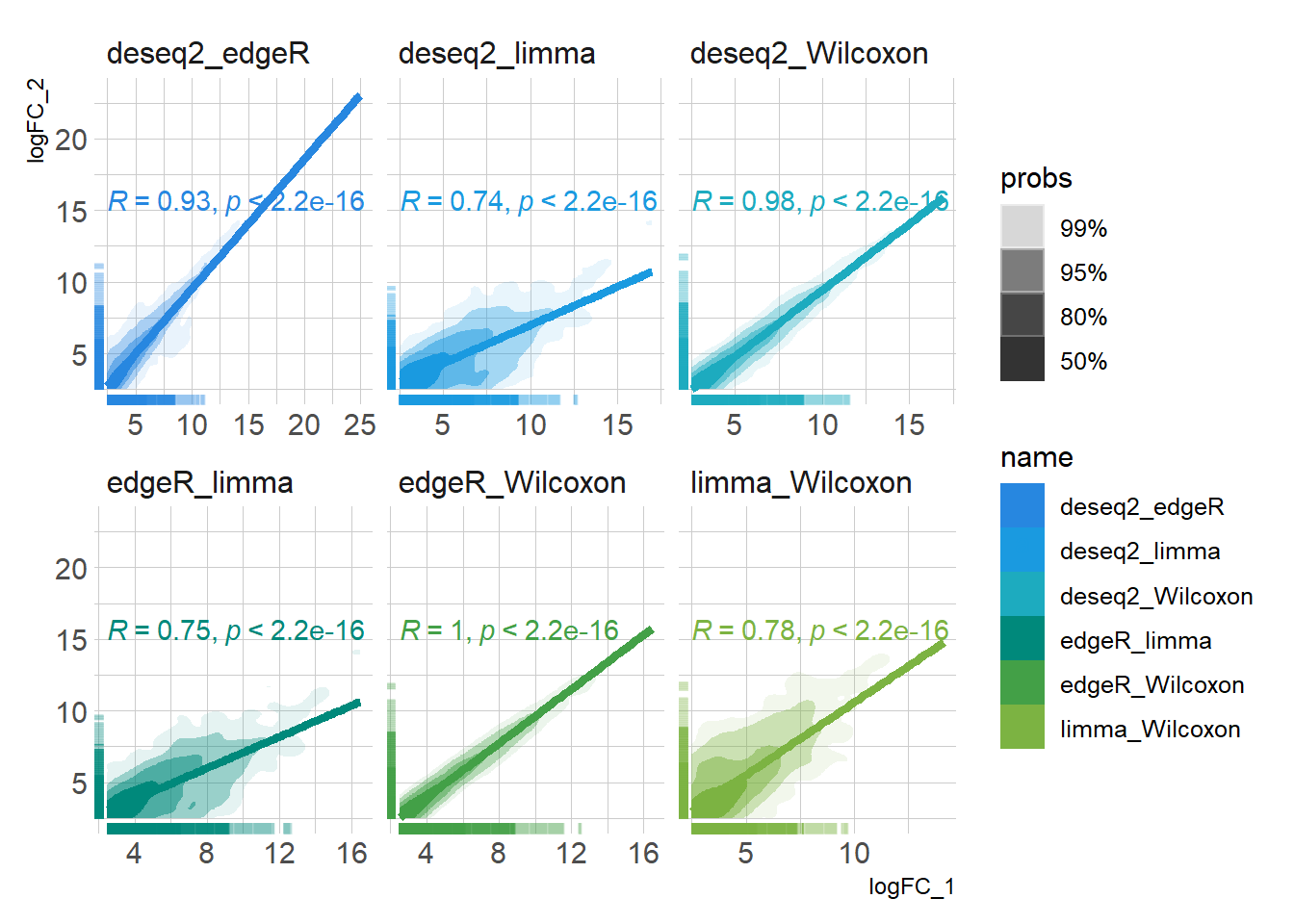
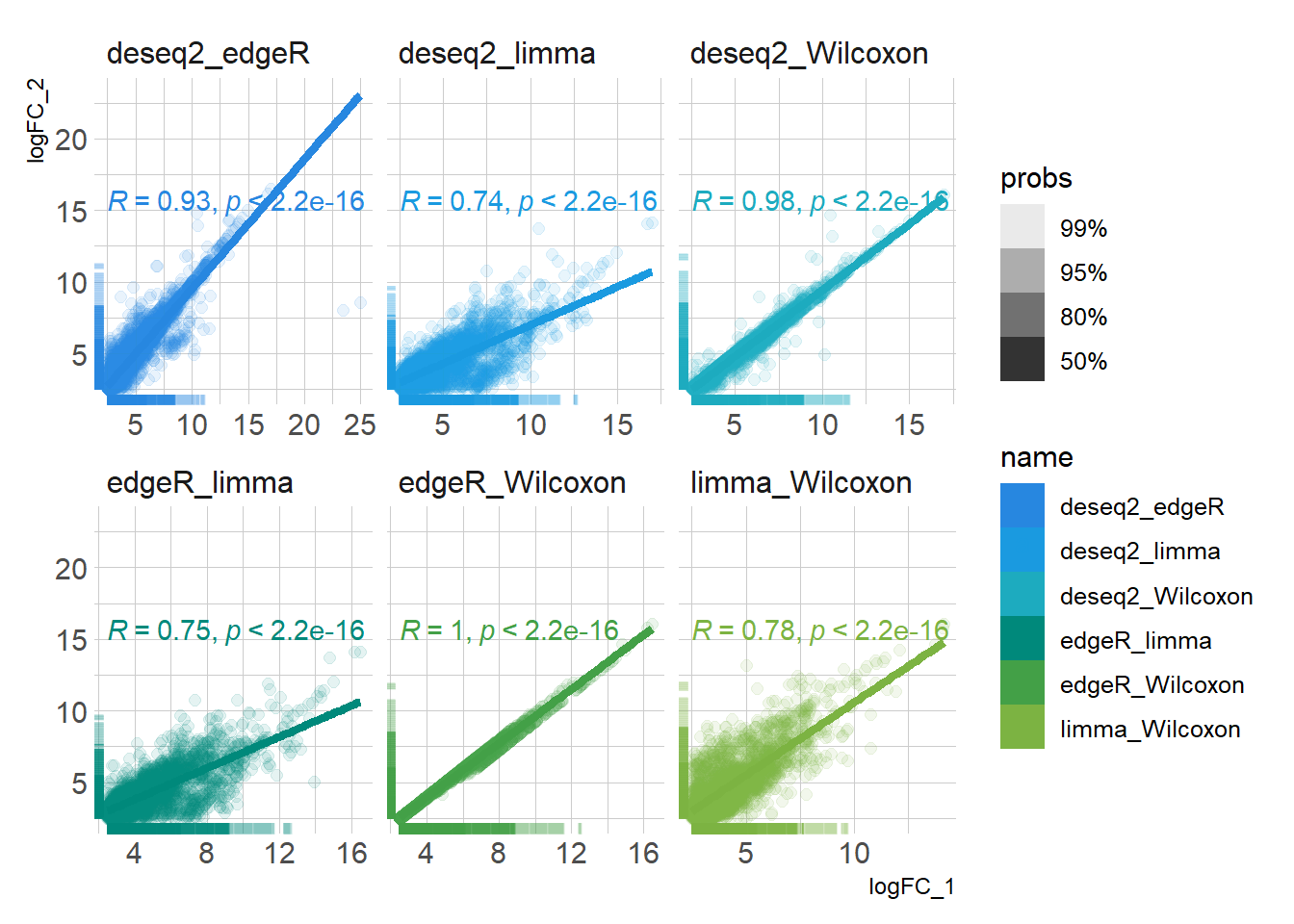
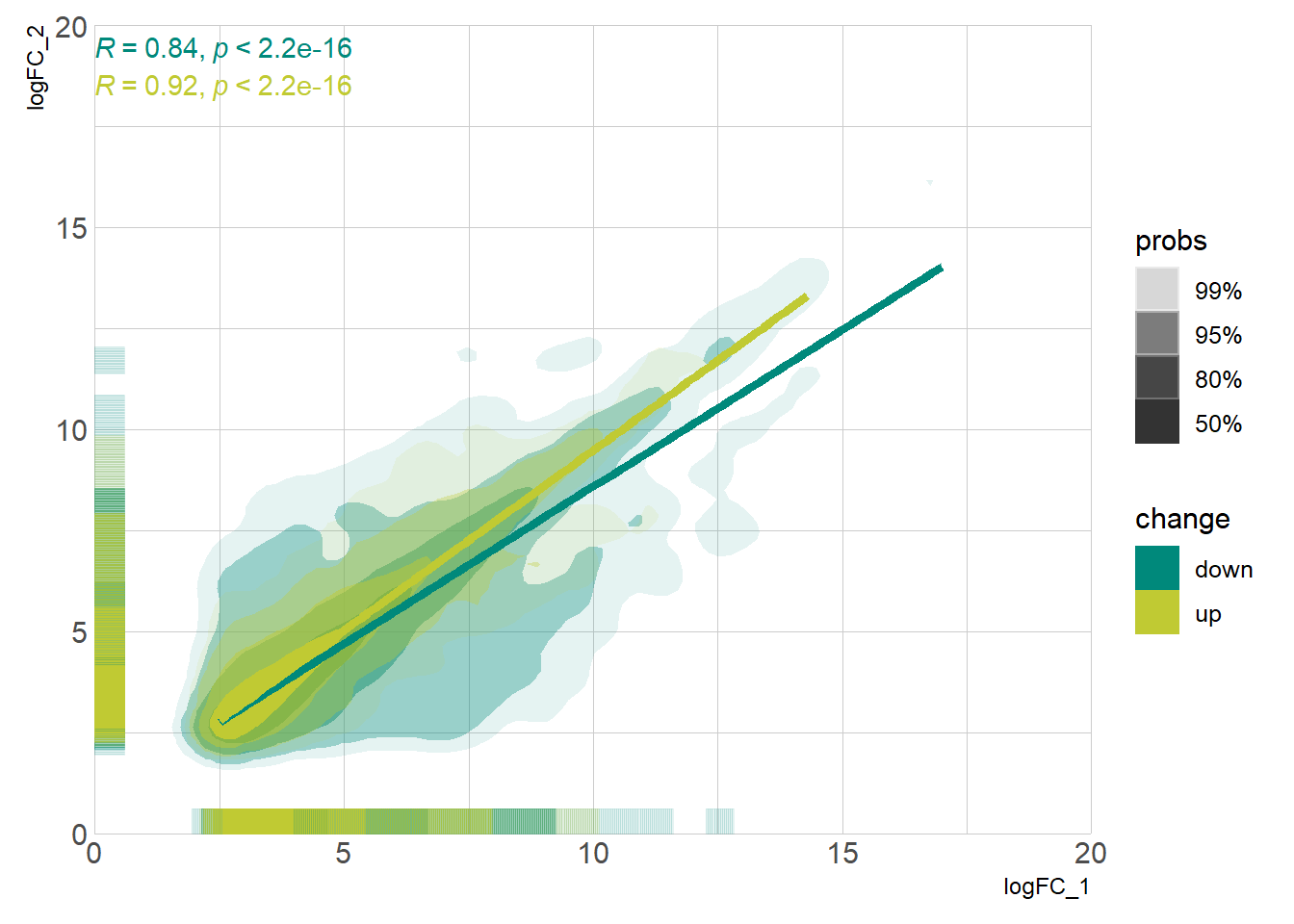
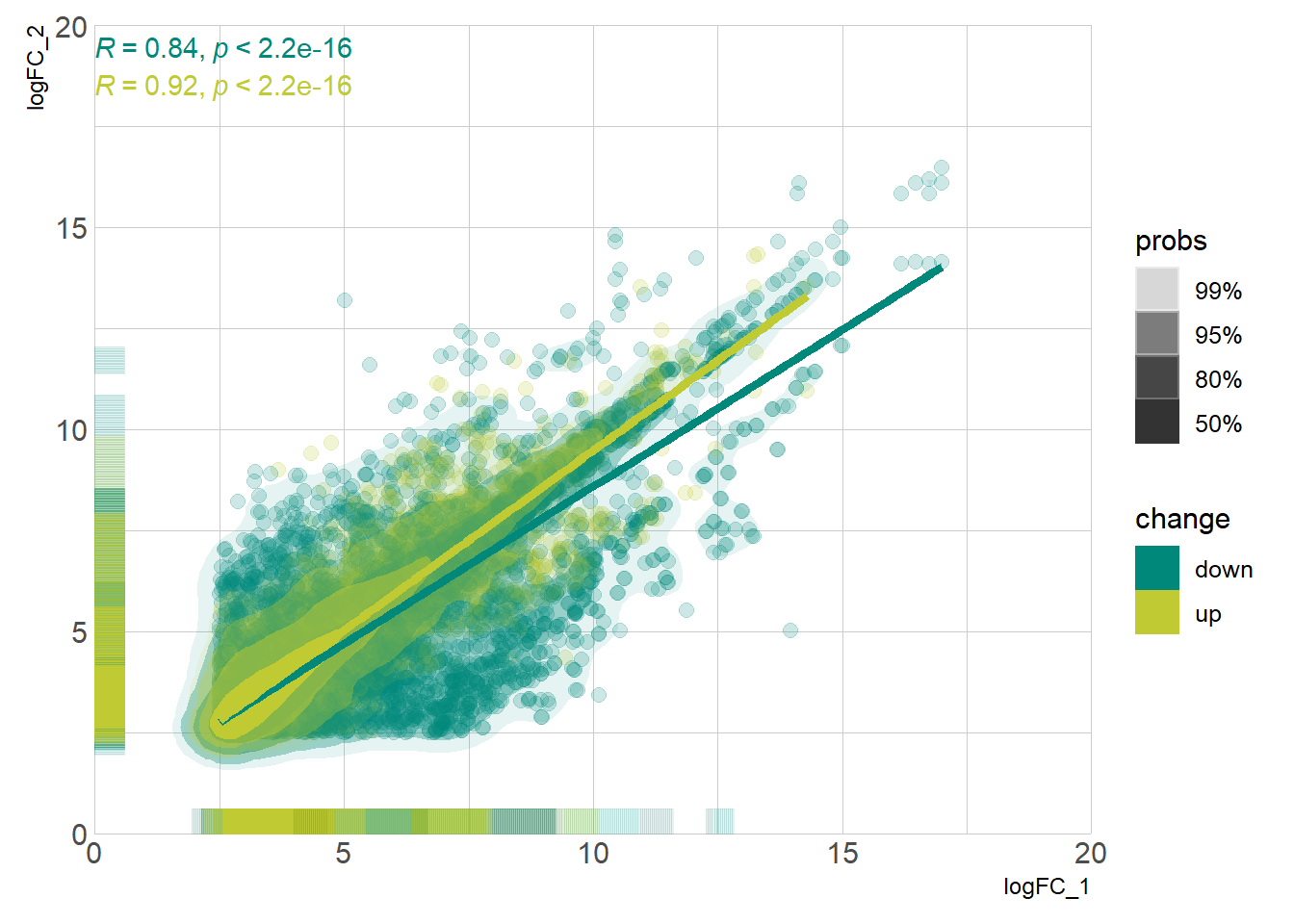
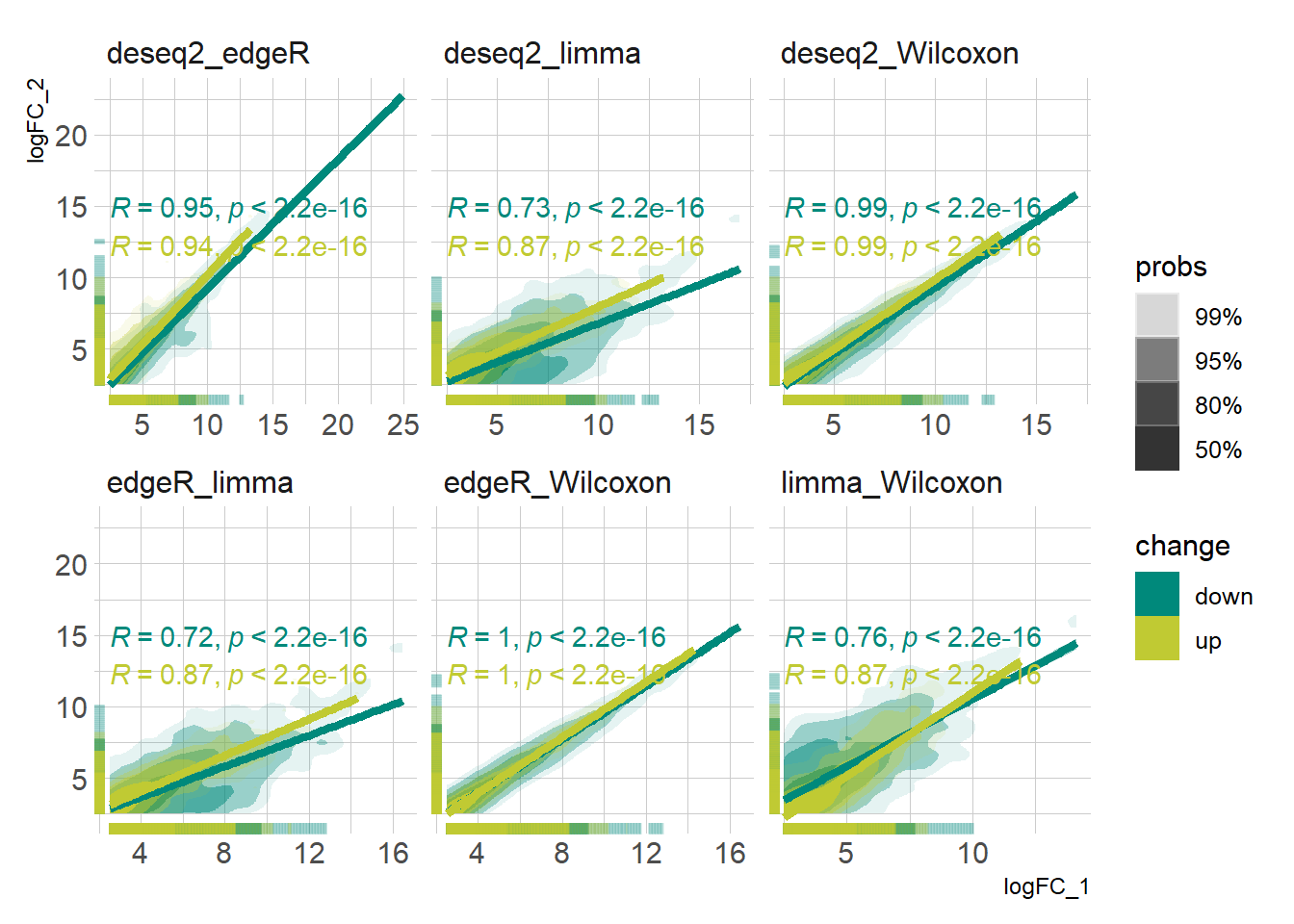
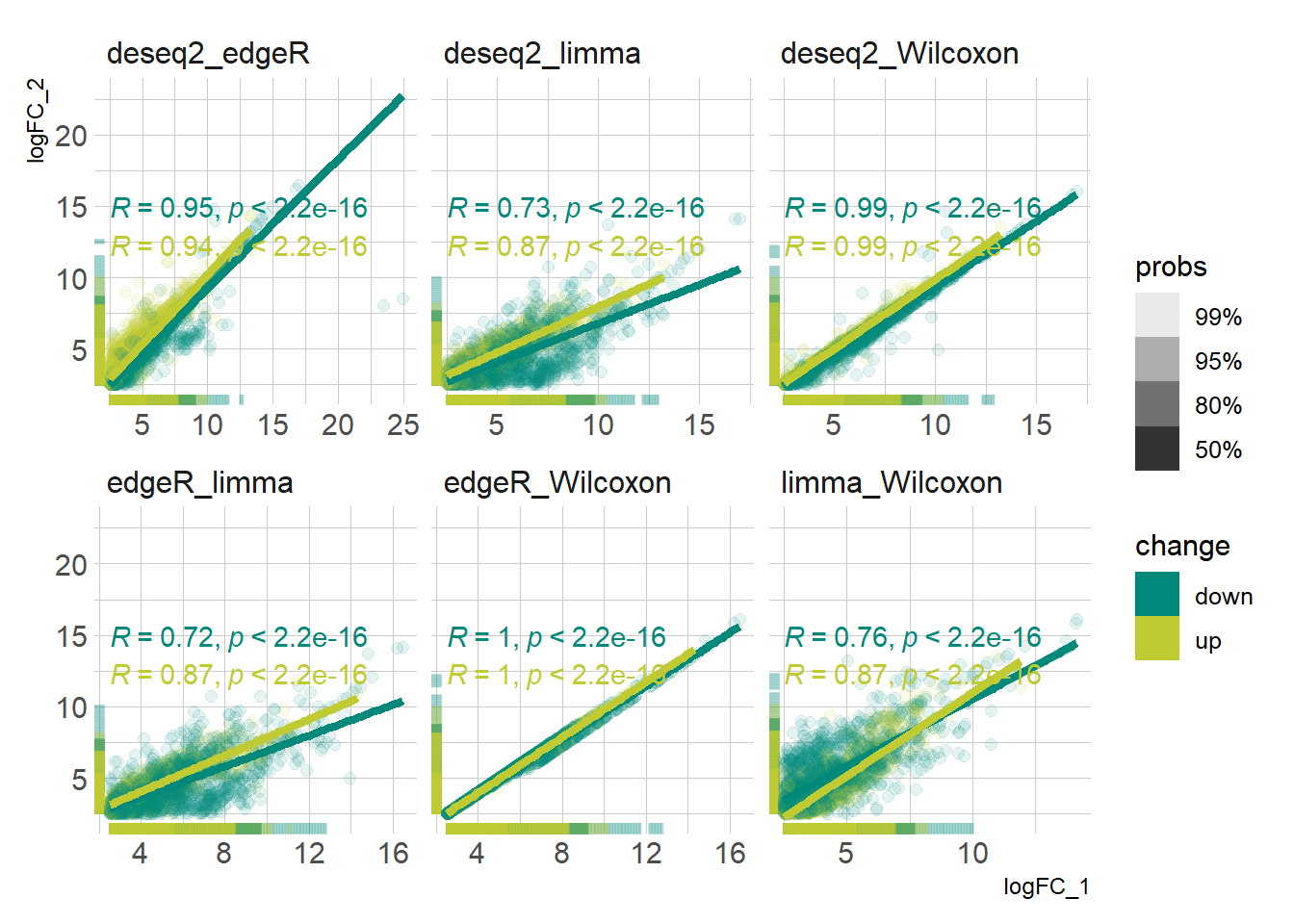
DEG_deseq2 <- prep_deseq2(
"../test_TransProR/Select DEGs/DEG_deseq2.Rdata"
)
head(DEG_deseq2, 5) logFC Pvalue change Gene
A1BG 2.549682 15.65356 down A1BG
A2M 3.251168 15.65356 up A2M
AADACL2 8.229956 15.65356 down AADACL2
ABCA12 6.392086 15.65356 down ABCA12
ABCB5 5.644108 15.65356 up ABCB5